In this article 
View / Download pdf
version of this article
Developed with the assistance of Dr Dorothy Saville - Senior Lecturer University of Otago, School of Pharmacy
Bioequivalence is defined as the absence of a significant difference in the rate and extent
of absorption into the systemic circulation, of two pharmaceutically equivalent medicines, when administered in the same
dose under similar conditions. Therapeutic effect (in terms of efficacy and safety) of bioequivalent medicines is considered
to be essentially the same.1
The rate and extent of absorption of an active ingredient in a medicine is defined as its bioavailability.
2 Pharmacological
response is related to the concentration of an active ingredient at the site of action (receptor site). Drug concentrations
cannot usually be measured at the site of action so it is assumed that the drug concentration at the receptor site is
in equilibrium with that in the blood. Most bioavailability studies therefore measure the drug concentration in blood.
The bioavailability of the active ingredient is what determines a product’s clinical efficacy.3
Bioavailability is measured using three main parameters - the area under the plasma drug concentration versus time curve
(AUC), the maximum plasma concentration (Cmax) and the time to reach maximum concentration (Tmax).
Bioequivalence can be determined by a comparison of the bioavailability of two formulations
of the same drug given at the same dose. The generic (or new brand) is always compared with the innovator (or reference)
product. Wherever possible, both products are tested in the same group of subjects in a randomised cross-over study. The
two medicines may be said to be bioequivalent if the 90% confidence intervals for the ratios of the geometric means (generic:innovator)
of the AUC and Cmax fall between 0.8 and 1.25 (80% and 125%). The Tmax of the generic and innovator
version of the drug must also be similar and there should not be a marked difference in inter-subject variability.2
In practice, the generic company tries to achieve a ratio of bioavailability (AUC,Cmax) close to 1. If the
ratio is closer to 0.8 or 1.25, then the data would have to be very uniform for the 90% confidence intervals of the ratios
to lie in the 0.8 to 1.25 range and therefore achieve bioequivalence.2
According to FDA guidelines for bioequivalence, a generic copy of a drug must contain identical amounts of the active
ingredient in the same dose formulation and route of administration. Some inactive ingredients (excipients) are allowed
to differ but must occur in a similar ratio to the active ingredient as that observed in the innovator drug.4
Simulation of a drug concentration versus time curve for two drug products
Adapted from Birkett D, 2003
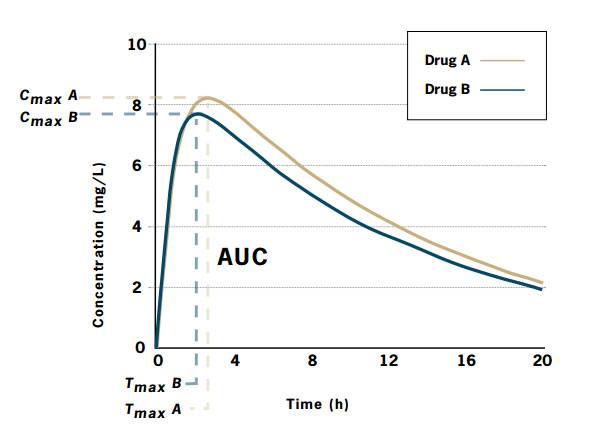 |
| Cmax |
maximum plasma drug concentration, Tmax time required to achieve a maximal concentration, |
| AUC |
total area under the plasma drug concentration-time curve |
Drug A is the innovator product and Drug B is the generic product.
| Drug A: |
Cmax = 8.1 mg/L; Tmax = 2.6 h; AUC = 124.9 mg.h/L |
| Drug B: |
Cmax = 7.6 mg/L; Tmax = 2.1 h; AUC = 112.4 mg.h/L |
The ratio of areas (generic:innovator), and therefore the relative bioavailability, is 0.9. To be accepted as bioequivalent,
the 90% confidence intervals for the area ratio would need to fall within the range 0.8-1.25.
How is bioequivalence regulated in NZ?
Adapted from Medsafe Bioequivalence Guidelines.1
In New Zealand, Medsafe is responsible for determining that a generic copy of a drug is bioequivalent to the innovator
version, before it is released onto the market. Medsafe bases bioequivalence testing guidelines on overseas regulations
and on what they regard as best current international practice.
Guidelines from the following regulatory authorities are currently used by Medsafe:
- European Commission Rules Governing Medicinal Products in the European Community Volume III and CPMP Notes for Guidance
- United States Food and Drug Administration (FDA)
- Australian Therapeutic Goods Administration (TGA)
- Therapeutic Products Directorate, Health Product and Food Branch, Health Canada
- World Health Organisation (WHO)
Any company wishing to manufacture or distribute a generic version of an innovator drug in New Zealand must submit
a Comparative Bioavailability Study report, in compliance with international standards, to be considered by Medsafe.
Variables included in a bioequivalence study
Ideally the bioavailability of systemic medicines should be measured using blood plasma or serum concentration of the
active ingredient. Where this is not possible, the quantity of the active ingredient or its metabolites excreted in urine,
or pharmacodynamic variables (e.g. heart rate) may be measured. However this results in a less accurate measure of bioavailability.
Single dose studies are appropriate in most cases. A steady-state study may be used in certain circumstances including;
medicines with a long terminal elimination half-life, highly toxic medicines, modified release products, medicines which
induce their own metabolism, enteric coated preparations (if coating is innovative), combination products, medicines that
exhibit non-linear pharmacokinetics and medicines which are likely to systemically accumulate.
Bioavailability studies are usually carried out in healthy adult human volunteers of both genders (where appropriate),
of average weight and between eighteen and sixty years of age. The number of subjects needed should be based on the number
required to reach statistical significance. The acceptable number of subjects is usually greater than twelve and less
than forty.
Experimental conditions should be standardised including gastrointestinal conditions, posture, physical activity and
timing of samples. The test formulation of tablets or capsules should originate from a batch of at least 10% of full production
scale or 100 000 units (whichever is greater) and should be manufactured using full production scale equipment. The mean
potencies (actual drug content) of the generic and innovator product should not differ by more than 5%.
What are the main issues with the validity of bioequivalence?
The introduction to the market of a generic drug, especially when replacing the innovator counterpart, is often met
with suspicion and concern by health care providers and patients. Concerns mainly surround the issue of bioequivalence
and whether use of the generic drug will result in unforeseen effects. Generic drugs are often perceived as being inferior
due to their lower cost and the lesser extent of development that goes into manufacturing these drugs compared to the
innovator version.5
The measure of bioequivalence
There has been some criticism of the use of the 80-125% reference range for bioequivalence in drugs which have a narrow
therapeutic range such as carbamazepine, phenytoin and digoxin.6 A relatively small change
in systemic concentration of these drugs can lead to a markedly different therapeutic response or even toxicity. Warfarin
also has a narrow therapeutic range and bioequivalence has not been established between the two main brands of this drug.
Therefore the two variants are not considered interchangeable.2 Similarly, concerns have
been raised over using this reference range for drugs with a wide therapeutic range, for example antibiotics and antihistamines.
5
Testing bioequivalence in a “normal and healthy” population
When an innovator drug is developed, evidence is required of its pharmacokinetics, efficacy and tolerability in volunteer
study subjects as well as the target population. However the development of a generic equivalent requires only evidence
of its bioequivalence with the innovator drug in the study subjects. This leaves some doubt as to whether the generic
drug would perform differently in a patient population, taking into consideration factors such as co-morbidities, concurrent
prescriptions and physiological factors such as differences in first pass metabolism, gastric pH and bacterial flora. 5
Older patients may also experience unique difficulties with a switch to a generic drug. Many suffer from multiple medical
conditions and receive multiple drugs which may affect pharmacokinetic properties. Physiological changes associated with
ageing may also affect drug absorption, distribution, metabolism and excretion.7 Bioequivalence
is generally tested in healthy subjects under the age of sixty.
Use of single-dose studies and the potential effect of excipients
Bioequivalence studies most often involve single doses of a drug.1 In clinical practice,
most drugs are administered in multiple doses and require maintenance of a steady-state. The maximum drug concentration
attained at a steady state is often higher than that achieved after a single dose.5 It is
possible that excipients used in the generic formulation (preservatives, pH adjusters, thickening agents etc) could affect
the absorption, and metabolism at steady state without producing these differences from a single dose.8 Excipients
can not always be considered inactive or inert.3 Some patients could have individual reactions
or sensitivity to a change in excipient.2 The potential effects of drug accumulation may
also not be seen with a single dose study.
Has the validity of bioequivalence been tested in NZ?
Adapted from Medsafe Media Release.9
In 2002 a drug company brought a challenge to the High Court against Medsafe’s procedures in evaluating the safety and
efficacy of a generic version of the drug, paroxetine mesylate. The company claimed that Medsafe had not followed its
own procedures for assessing the generic drug and that clinical trials may be required to confirm that paroxetine mesylate
was safe and effective.
The court case reviewed Medsafe’s handling of the process for approval of the generic drug. This was supported by chemical,
pharmaceutical and bioequivalence data which established the product’s quality, safety and efficacy. The data showed that
the generic version of paroxetine did not solely rely upon pre-existing toxicological data for the innovator product.
The data also demonstrated bioequivalence between the generic and innovator product, with respect to the same amount of
active substance being absorbed to the same extent.
The High Court rejected all grounds of challenge by the drug company and found in favour of Medsafe. It was ruled that
the evaluation process for the generic drug was robust and followed correctly and that Medsafe properly considered all
information about the drug.
The outcome of this challenge can provide reassurance that Medsafe applies rigorous procedures to evaluate the safety
and quality of medicines before they are made available to the public.
So what does this mean?
There is no recent documented evidence of proven failure of a generic formulation of a drug, due to issues of bioequivalence.
There are some reports of therapeutic inequivalence, however most of these cases were determined to likely be the result
of progression of disease rather than lack of bioequivalence of a generic and innovator formulation of a drug.10
Given the fact that distributors of generic drugs in New Zealand must provide scientific evidence of bioequivalence
in accordance with Medsafe’s guidelines, it can be assumed that if a generic drug is on the market, it can be considered
therapeutically equivalent to the innovator counterpart, unless classified as non-interchangeable.
Bioequivalence of Loxamine vs Aropax
Can Loxamine and Aropax be considered bioequivalent?
Yes, information received from Pacific Pharmaceuticals shows that the results of the studies on Loxamine are well within
the bioequivalence acceptance limits. This means that any variation in bioavailability (AUC, Cmax) between
Loxamine and Aropax is very unlikely to be any different from variations between different batches of the same brand.
| Loxamine vs Aropax: Bioequivalence study results |
 |
| 90% confidence intervals for ratios of geometric means |
Cmax
Tmax
AUC 0–t
AUC 0–infinity |
0.978 to 1.117
0.968 to 1.020
1.000 to 1.139
0.998 to 1.136 |
| Loxamine and Aropax are bioequivalent (90% CI’s within 0.8–1.25) |
Loxamine vs Aropax: Inactive ingredients (excipients)
Both tablets contain 20 mg paroxetine hydrochloride
Aropax tablets also contain; the colouring agent titanium dioxide (white, E171), calcium hydrogen phosphate, hypromellose,
sodium starch glycolate (potato starch), magnesium stearate, polysorbate 80 and macrogol 400.
Loxamine tablets also contain; calcium hydrogen phosphate anhydrous, sodium starch glycolate, colloidal anhydrous silica,
magnesium stearate, purified talc, titanium dioxide and Eudragit 100.
Loxamine contains silica, talc and Eudragit 100 which are not present in Aropax tablets. Silica and talc are widely
used, relatively inert compounds used as tablet fillers. Eudragit 100 is a polymethacrylate which is extensively used
as tablet film coating. These agents are selected on the basis that they are very unlikely to cause adverse effects but
the remote possibility of sensitivity to these excipients cannot be completely excluded.
References
- Medsafe. New Zealand regulatory guideline for medicines. Section 15. Bioequivalence testing of oral medicines, 2001;1.
Available from
http://www.medsafe.govt.nz/regulatory/Guideline/NZRGM
Volume 1.asp. Accessed February 2007.
- Birkett D. Generics - equal or not? Aust Prescr, 2003;26:85-7. Available from
http://www.australianprescriber.com/magazine/26/4/85/7.
Accessed February 2007.
- Borgherini G. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther,
2003;25:1578-92.
- US Food and Drug Administration. Bioavailability and bioequivalence requirements. Fed Regist, 1992;57:17997-18001.
Available from http://www.fda.gov/cder/ogd/. Accessed February
2007.
- Meredith P. Bioequivalence and other unresolved issues in generic drug substitution. Clin Ther, 2003;25:2875-90.
- Reiffel J. Issues in the use of generic antiarrhythmic drugs. Curr Opin Cardiol, 2001;16:23-29.
- Gerbino P, Joseph A. Multisource drugs: Implications and concerns in the geriatric population. Hosp Pharm, 1993;28:96-102.
- Besag F. Is generic prescribing acceptable in epilepsy? Drug Saf, 2000;23:173-182.
- Medsafe. Paroxetine mesylate - High Court rejects challenge. Media Releases, 2002. Available from
http://www.medsafe.govt.nz/hot/media/media2002.asp.
Accessed February 2007.
- Rheinstein P. Therapeutic inequivalence. Drug Saf, 1990;5(Suppl 1):114-119.