Flecainide is a Class I antiarrhythmic medicine, therefore it slows cardiac contraction by blocking sodium channels.1 Flecainide
is typically initiated in secondary care for rhythm control in people with atrial fibrillation but it is also used for
other arrythmias (for details see: nzf.org.nz/nzf_1096).2 Prescriptions of
flecainide need to be endorsed by a specialist (including vocationally registered general practitioners). Flecainide is
usually initiated at a low dose which is increased according to clinical response with close monitoring of the patient
until the maintenance dose is achieved.
The funded brands of controlled and immediate release flecainide are changing

Previously, the only brand of flecainide controlled release capsules (100 mg and 200 mg) that was funded in New Zealand
was Tambocor CR. There are approximately 6,200 patients taking flecainide controlled release capsules in New Zealand.3
- From 1 July, 2019, Flecainide (Teva) controlled release 100 mg and 200 mg capsules are funded and will replace
Tambocor CR over a five-month transition period.4
- From 1 December, 2019, Flecainide (Teva) will be the only funded brand of controlled release flecainide (until
at least 30 June, 2022) and Tambocor CR will not be funded.
Currently, the only brand of flecainide immediate release tablets (50 mg) that is funded in New Zealand is Tambocor.
There are approximately 1,600 patients taking flecainide immediate release tablets in New Zealand.
- From 1 September, 2019, Flecainide BNM 50 mg immediate release tablets will be funded and will replace Tambocor over
a five-month transition period.4
- From 1 February, 2020, Flecainide BNM will be the only funded brand of flecainide 50 mg tablets (until at
least 30 June, 2022) and Tambocor will not be subsidised.
Flecainide immediate release is occasionally extemporaneously compounded as an oral liquid; PHARMAC is currently working
with the manufacturer of Flecainide BNM and stakeholders to ensure that this is possible.4
A transition period of five months is provided for both flecainide brand changes to allow patients and health professionals
adequate time to discuss the change and convert to the new brand.
Plasma and ECG monitoring are not required during a brand change
Patients taking flecainide are monitored frequently by ECG when treatment is initiated and when doses are increased.1 Plasma
concentration monitoring may also be beneficial for patients with renal or hepatic dysfunction to guide titration of dosing.
ECG and plasma monitoring are not required during a flecainide brand change unless the patient is displaying cardiac symptoms
or signs that suggest a dose adjustment may be necessary.4 If the treating clinician determines that plasma
monitoring is advisable, an application to PHARMAC can be made to cover the patient co-payment of one additional consultation
per patient for plasma monitoring. Practices can invoice PHARMAC for the General Practitioner co-payment fee in lieu of
charging the patient.
Further information on invoicing for the consultation fee associated with flecainide plasma monitoring
is available from:
https://pharmac.govt.nz/medicine-funding-and-supply/medicine-notices/flecainide/#bookmark3
(See: “For healthcare professionals”)
A brand switch fee is available for pharmacists
Community pharmacists also need to provide education and reassurance to people when they change to a different brand
of medicine. It will be necessary to stock all subsidised brands of flecainide during the transition period. Community
pharmacies will be able to claim a brand switch fee to compensate them for any additional counselling required to successfully
transition patients on the first dispensing of the relevant medicine during the following periods:4
1 December, 2019 to 1 March, 2020 – a brand switch fee will apply for controlled release flecainide capsules
1 February, 2020 to 1 May, 2020 – a brand switch fee will apply for immediate release flecainide tablets
Reassure patients that the active ingredient is unchanged
Medsafe has approved Flecainide (Teva) using Tambocor CR as the reference medicine and flecainide BNM has been approved
with Tambocor as the reference medicine. This means that the newly funded medicines contain the same active ingredient
in the same quantity and bioequivalence has been demonstrated with respect to flecainide (see: “Bioequivalence is used
to establish the safety of generic medicines“). The only known difference between the newly funded flecainide and the
soon to be delisted formulations is that immediate release flecainide BNM does not contain hydrogenated vegetable oil.4
There is little difference in the appearance between the newly funded brands of flecainide (Figure 1 – right) and the
brands of flecainide that will be delisted (Figure 1 – left).
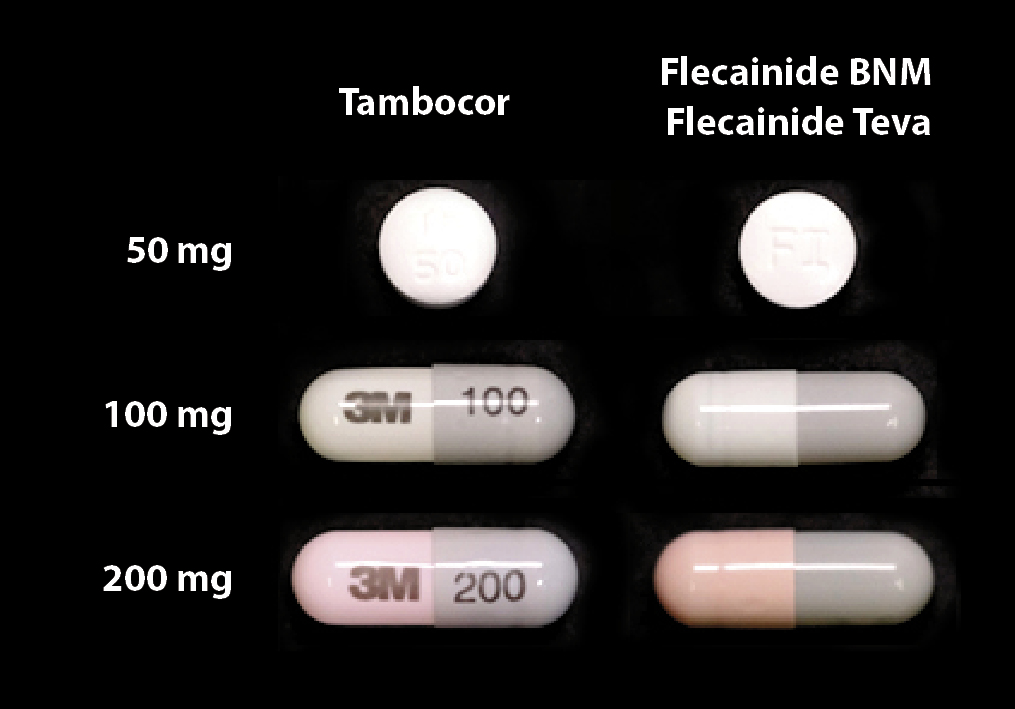
Figure 1:Tambocor immediate release flecainide (50 mg)
and Tambocor CR flecainide (100 mg and 200 mg) on
the left (soon to be delisted) and newly funded Flecainide BNM (50 mg) and Flecainide (Teva – 100 mg and 200 mg) on the right
Information for patients taking controlled release flecainide information is available from:
https://pharmac.govt.nz/assets/2019-flecainide-brand-change-pamphlet-english.pdf
Amiodarone is classified as a Class III antiarrhythmic medicine as it blocks potassium channels. Amiodarone is typically
initiated in secondary care for patients with atrial fibrillation and other tachycardias (for details see:
nzf.org.nz/nzf_1090).2 Prescriptions
of amiodarone need to be endorsed by a specialist (including vocationally registered general practitioners).
Amiodarone is associated with serious adverse effects including pulmonary and hepatic toxicity and thyroid dysfunction.6 Regular
monitoring in primary care is therefore necessary,6 however, additional monitoring is not generally required
when patients change brands of amiodarone.
Further information on monitoring patients taking it is available from:
www.saferx.co.nz/assets/Documents/ee2548bd3c/amiodarone.pdf
The funded brand of amiodarone is changing
Previously, the only brand of amiodarone capsules (100 mg and 200 mg) that was funded in New Zealand was Cordarone-X.
There are approximately 6,900 patients taking amiodarone capsules in New Zealand.3
- From July 1, 2019, Aratac 100 mg and 200 mg amiodarone tablets are funded and will replace Cordarone-X over
a five-month transition period.7
- From 1 December, 2019, Aratac will be the only funded brand of amiodarone and Cordarone-X will not be funded.
A transition period of five months is provided to allow patients and health professionals adequate time to discuss the
change and convert to the new brand. This brand change is a reversal of one that occurred in January, 2017, when Cordarone-X
replaced Aratac as the only funded brand of amiodarone tablets.8 Patients who have been taking amiodarone
for a number of years will therefore be changing back to the medicine they were originally taking. A brand switch fee
will not be available to community pharmacists for the amiodarone transition.
Information for patients
There is very little difference in the appearance of the two brands of amiodarone other than differences in markings
and the 100 Aratac mg tablets are slightly bigger than the 100 mg Cordarone-X tablets (Figure 2).
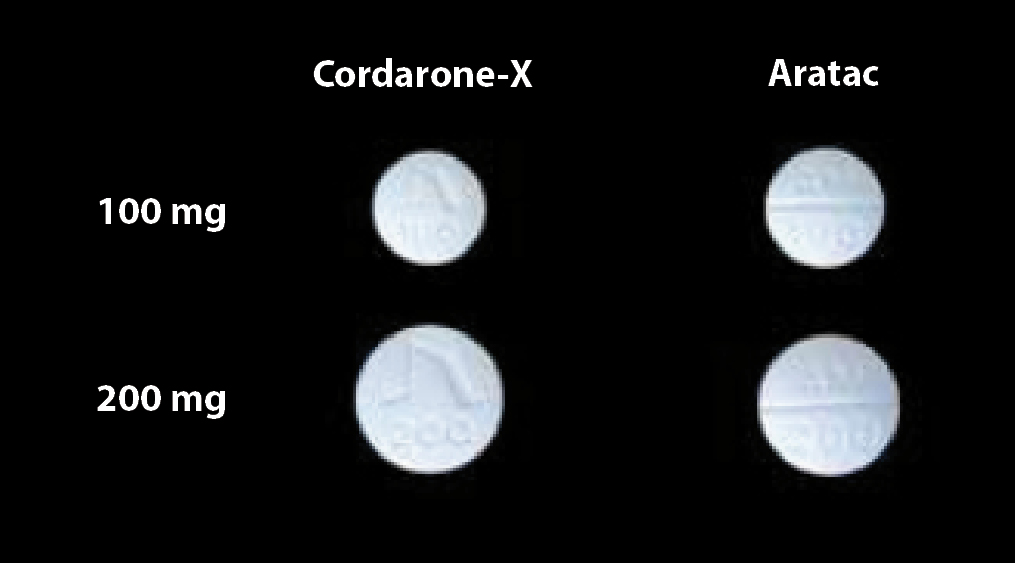
Figure 2: Cordarone-X (100 mg and 200 mg amiodarone) on the left
(soon to be delisted) and Aratac (100 mg and 200 mg amiodarone) on the right
A brand change is traditionally thought of as changing to a generic medicine (see below) from an original brand, also
referred to as the innovator medicine. However, medicine brands may also be changed from generic to generic and from generic
to innovator. A strict regulatory framework exists in New Zealand to ensure that the quality and supply of any medicines
undergoing a brand change is not compromised.
A generic medicine is required to be bioequivalent to the reference medicine which is usually the innovator brand. Generic
medicines cannot be sold until the patent on the innovator medicine has expired. The generic medicines that are approved
for use in New Zealand by Medsafe are manufactured according to the same international standards and Good Manufacturing
Practice as innovator medicines.9
Further information on generic medicines for patients is available from:
https://pharmac.govt.nz/medicine-funding-and-supply/what-you-need-to-know-about-medicines/generic-medicines/
Adverse reactions to excipients are extremely rare
Excipients are substances included in medicines to aid in manufacturing, to protect or enhance the stability of the
active ingredient or to improve bioavailability or patient acceptability.10 Manufacturers must supply Medsafe
with a list of all the excipients used in their medicines to ensure they are not toxic, are internationally approved and
that the risk of adverse reactions is minimal. It is, however, possible for patients to experience an adverse reaction to an excipient.
All the excipients contained in medicines are listed on the medicine’s data sheet, available from:
www.medsafe.govt.nz
Ask patients to report any concerns about adverse effects
Adverse effects following a brand change are highly unlikely, however, patients should be advised to report any concerning
symptoms. Regarding the flecainide and amiodarone brand changes, if patients report cardiac symptoms, an ECG may provide
reassurance to patients and can also detect changes in their condition that are unrelated to the brand change. This is
also an opportune time to reinforce to patients taking amiodarone the importance of reporting respiratory symptoms early,
e.g. unexplained cough or dyspnoea, as adverse pulmonary effects usually require treatment withdrawal.
Bioequivalence is used to establish the safety of generic medicines
Clinical trials to test the efficacy of the active ingredient of generic medicines are not required as these have already
been conducted by the manufacturers of the innovator brands. Manufacturers of generic medicines are instead required to
present clinical evidence to Medsafe to demonstrate that the medicine is bioequivalent to the originator brand of medicine
or a suitable alternative. Once bioequivalence is demonstrated it is assumed that the medicine will have the same clinical
effects.9
Bioequivalence studies measure aspects of absorption and blood levels of the active ingredient in no less than 12 healthy
volunteers.11 This includes the parameter ‘area under the curve’ (AUC), which reflects the extent and duration
of exposure to the active ingredient, and the parameter maximum plasma concentration (Cmax).11 It
is assumed that if the generic and innovator medicine behaves the same way in healthy people, they will also behave the
same way in people with morbidities. The study protocol is randomised and has a cross-over design with a washout period,
so each participant is administered the generic and innovator medicine.9
Bioequivalence is established if the 90% confidence interval for the ratio of the generic and reference medicines is
within the range of 80–125%.11 The actual difference in exposure to the active ingredient between generics
and innovators is less than 5%.9 The 80–125% acceptance range is the international standard for demonstrating
bioequivalence.11 This range accounts for statistical error and is generally considered to be clinically
insignificant.9 If a medicine has a narrow therapeutic range, i.e. there is only a small difference between
the effective dose and doses which produce unacceptable adverse effects, the acceptance range for bioequivalence reduces
to 90–111%.9 N.B. Flecainide is an example of a medicine that is considered to have a narrow therapeutic
range.12
The appropriateness of using bioequivalence as a surrogate for clinical efficacy and safety is supported by numerous
studies. A systematic review of 74 studies investigating the efficacy of generic cardiovascular medicines in comparison
to branded medicines found no significant difference in clinical outcomes.14
Further information on bioequivalence and the approval process for generic medicines is available at:
https://medsafe.govt.nz/profs/PUArticles/Mar2013GenericMedBioqueivalence.htm and
www.medsafe.govt.nz/profs/PUArticles/September2017/TheMedsafeFiles4NMAssessment.htm






